Biological Proteins: Amino acids, peptide bonds, and protein structures
Did you know that the human body contains an estimated 100,000 different proteins, all due to the numerous ways that only 20 amino acids can combine? Proteins are large molecules made up of hundreds, even thousands, of amino acids combined in different ways.
Spider silk, hemoglobin, keratin in your nails and hair, actin and myosin in muscle fibers – all these are proteins. As a class of biological compounds, they are vital to essentially every biological process, because they can take so many different forms. Proteins can be long fibers with the ability to slide as in muscles; they can be large and globular, like von Willebrand factor which helps in blood clotting; or they can be small like insulin, which is needed for sugar metabolism. Insulin is one of the most well-known proteins because of its use to treat diabetes, but it is also familiar to biochemists because it was the first complete protein structure discovered.
In 1921, Frederick Banting and Charles Best extracted insulin from the pancreas of dogs and learned that it was a hormone affecting blood sugar levels. Within a year, it was used to save the life of a diabetic boy. This set off a wave of research that put insulin at center stage, peaking in the 1950s when British biochemist Frederick Sanger figured out the precise sequence by which the amino acid building blocks are put together to build insulin.
The building block sequence
During World War II, when Sanger turned his attention to insulin, he and other biochemists of the era already knew that this hormone was a protein. Today, we know that proteins are polymers composed of building blocks called amino acids (Figure 1).
Figure 1: The general structure of an amino acid.
A multitude of amino acids are possible. In fact, the Murchison meteorite (Figure 2), which fell in Australia in 1969, was found to contain seventy different amino acids, but life on Earth uses just twenty, but that’s enough to create an astronomical number of possible proteins.
Figure 2: The Murchison meteorite, which landed in Australia in 1969, has been shown to contain many types of chemicals required by life on Earth. On the right is a pebble-sized fragment of the meteorite; when magnified 10 times and placed in polarized light, a slice of the meteorite reveals various minerals in different colors.
image ©NASAThe human body alone contains an estimated 100,000 different proteins, because of the numerous ways that the same 20 amino acids can combine. But scientists back in the early 20th century did not think that proteins were structured in any way that affected their function and Sanger was key to changing that idea.
Prior to Sanger’s major discoveries, biochemists learned about a feature in proteins called a disulfide bridge (Figure 3). They also found that treatment with chemicals called reducing agents severs a disulfide bridge between two cysteines and also causes a large proteins to split into smaller proteins, arguing that these bonds exist in proteins to help hold them together.
Figure 3: A disulfide bridge (the joined S molecules) connecting two cysteines.
Consequently, biochemists in the World War II era believed that amino acids must be linked in chains in a way that today we might liken to beads on a string. They knew that each amino acid in a chain was connected to the next amino acid through a special type of chemical connection called an amide bond, also called a peptide bond.
Comprehension Checkpoint
The peptide bond
To understand a peptide bond, we need to look more closely at the structure of amino acids. As noted earlier, the different types of amino acids are distinguished based on the R group. If R is a hydrogen atom, for instance, the amino acid is glycine. If R is a methyl group (CH3), the amino acid is alanine. If R is the sulfhydryl (CH2SH), the amino acid is cysteine. These are just a few examples, but apart from the R group all amino acids are otherwise the same. At one end, each amino acid has the functional group COOH, called carboxyl. At the other end, each amino acid has an NH2 group, called amino. (See Figure 4 for a peptide bond in an amino acid.)
Figure 4: Peptide bond
A peptide bond is formed when the carboxyl carbon atom of one amino acid is joined covalently with the amino nitrogen atom of another amino acid, expelling a molecule of water (H2O). Linking of several amino acids by their carboxyl and amino groups produces a small protein, also called a polypeptide, because it contains several peptide bonds (Figure 5). Joining amino acids in this way produces a chain with a COOH at one end and an NH2 at the other end, called the carboxyl and amino ends, respectively.
Figure 5: The joining of two amino acids (red) with a molecule of water expelled (blue).
By Sanger’s time, biochemists were using acidic chemicals to break the peptide bonds of a protein, thus separating the individual amino acids. Additionally, they knew that a protein could have more than one polypeptide chain, connected by another by disulfide bonds attaching at areas of a chain that contained cysteine. By treating a protein to destroy disulfide bridges, biochemists in the early 1940s could find out the number of chains in a protein. Also, by breaking apart the peptide bonds and running chemical tests, they could determine the identity of the amino acids of a protein and the relative amounts of each amino acid.
However, this did not tell biochemists the sequence in which those amino acids had been linked together. What set Sanger apart from his contemporaries was an insight that the relative amounts of each type of amino acid and their sequence could be extremely important. It might be the basis of how each protein functioned. If so, then amino acid sequence would also be the key to how life functioned. Given the prevalence of proteins in organisms, the idea made a lot of sense, but now Sanger’s task was to prove it. Doing this would be no easy task, but the first step was to choose a particular protein on which to concentrate his work.
Comprehension Checkpoint
Insulin as a model for the study of protein structure
Because it is small and important in the treatment of a disease, insulin was a logical choice for Sanger to begin his work on amino acid sequencing. He began with bovine insulin, since it was easy to obtain and purify in large quantities. The first thing he did was to treat the insulin with the chemical agent that broke up disulfide bridges. If insulin consisted of just one polypeptide chain, testing the size of the protein before and after chemical treatment would give the same result.
The amino acids in proteins carry electric charges, so a protein, or fragments of a protein, could be propelled in an electromagnetic field with different degrees of strength. This technique is called electrophoresis (Figure 6). It was very new in Sanger’s time but it gave him very clear results. Whereas prior to the disulfide bridge treatment, the insulin behaved in one particular way in electrophoresis, after the treatment, the electrophoresis produced two different results, both different from the pre-treatment result. This meant that the insulin had been divided into two sections, each with a slightly different size. In other words, the insulin consisted of two peptide chains and the task now was to find the amino acid sequence of each.
Figure 6: A modern example of gel electrophoresis. The laboratory set-up uses an electric current to separate molecules based on size.
image ©Jean-Etienne PoirrierJust as large fragments of a protein can be propelled in a particular way by electrophoresis, so can smaller fragments, including when a protein is fragmented down to pieces consisting of 10-15 amino acids each. He did the fragmentation by treating each chain with an enzyme called trypsin, which cuts only next to certain amino acids (lysine and arginine). Subsequently, he could utilize other enzymes to fragment each fragment more, all the way down to individual amino acids. Each fragment has its own pattern in electrophoresis.
With another technique called chromatography (Figure 7), Sanger could identify fragments that were bound to a certain chemical agent that he developed, known as dinitrofluorobenzene (DNFB), which could react chemically with amino groups that were not part of a peptide bond. After performing the first fragmentation using trypsin, but prior to fragmenting each piece further into individual amino acids, he added the DNFB, which altered whichever amino acid was at the amino end of the fragment (also called the N-terminal amino acid). Because of this, when he then broke the fragment into individual amino acids, the amino acid that had been at the N-terminal remained bound to the DNFB. He could identify this DNFB-bound amino acid in chromatography by comparing the chronographic signal of the broken down chain to 20 “standards” –samples of compounds consisting of DNFB bound to one of the 20 amino acids, each of which produced a distinct chromatography pattern.
Figure 7: A page from Frederick Sanger's notebook, detailing work on cow and pig insulin. On the right is one of his paper chromatograms.
image ©Frederick Sanger Papers, SABIO/P/1/13, Wellcome LibraryIf an amino acid had been altered with DNFB, that would be the amino acid at the amino end of the protein chain fragment. Knowing the identity of the amino acid at the N-terminal of the fragment, he could use an enzyme that would cut on the carboxyl end of that known amino acid, thereby producing a fragment featuring the next amino acid as the N-terminal amino acid. On that altered fragment, he could repeat the DNFB binding procedure and chromatography and this way learn the identity of that second amino acid of the fragment.
He repeated the technique for each fragment, thereby obtaining the amino acid sequence of all of them. Then, he repeated the entire procedure using an enzyme other than trypsin to break up the big chain into fragments of 10-15 amino acids, and then again using still a different enzyme. He used four different enzymes, each of which worked by cutting next to certain amino acids and not others, and this allowed only one possibility for the order in the fragments had been linked together in the original chain.
It was a long, tedious process, but Sanger had the amino acid sequence of the both chains in 1952. After another three years of similar chemical tactics, he and several coworkers demonstrated that for the insulin chains A and B to work together as physiologically functional insulin they had to be linked by three disulfide bridges at three distinct points (Figure 8).
Figure 8: F. Sanger's method of analyzing peptide end-groups. It begins with using his reagent, DNFB, to react with the N-terminal amino acid. The amino acid then remains bound to the DNFB (A). Through hydrolysis (B) he could then identify the amino acid using chromatography.
Insulin is considered small for a protein because together its two chains contain just 51 amino acids, but Sanger’s discovery applied to proteins overall. Small or big, proteins were built of specific amino acid sequences; changing the sequence would make it a different protein. The discovery that earned Sanger his first Nobel Prize in Chemistry in 1958. He would later earn a second Nobel Prize in chemistry for working out a similar approach for the sequencing of DNA, putting him on a very short list of people who have won the Nobel Prize more than once.
Comprehension Checkpoint
Sequences of amino acids influence the chemical properties of a protein
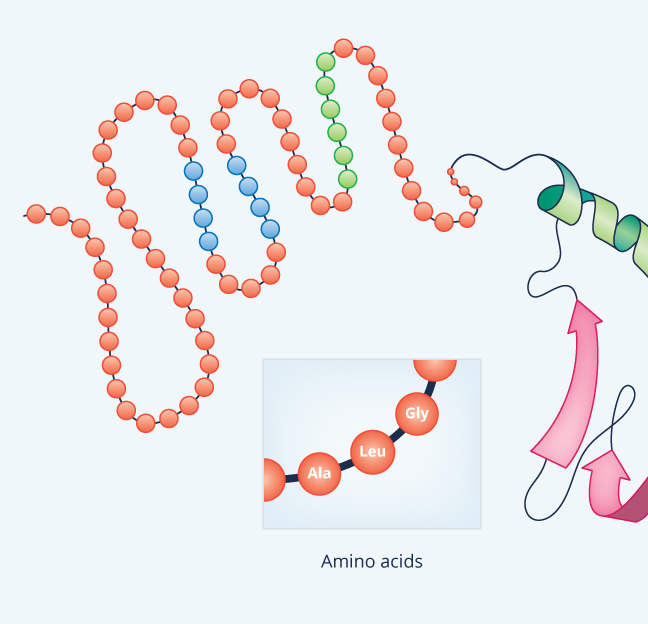
Sanger’s discovery with insulin revealed not just how proteins have defined chemical structures, but also why different proteins have different functions. Just as different letters of the alphabet have different sounds, the various R chains give the twenty amino acids different chemical properties. Thus, stringing amino acids together in different combinations leads to proteins with extremely diverse properties and shapes.
Sanger’s insulin research acted as a springboard for work by other protein chemists during the 1950s and 60s involving how structure relates to function. By passing X-rays through various proteins, researchers obtained images of their 3-dimensional structures. Studying the images and working out issues related to the physics of chemical bonds, biochemists of the mid 20th century learned that the amino acid sequence represents protein structure on just one level. They started referring to the sequence as the primary structure, since it leads the protein chain to twist and bend in ways that give the protein a more complex shape.
Certain amino acids enable a polypeptide chain to bend, for example, while other amino acids hold the chain more rigid (Figure 9). Some R chains are very hydrophilic; they like being in water and thus make the amino acid water-soluble. Other R chains are hydrophobic; they don’t mix with water. Often, having a hydrophobic area, or “pocket”, within a protein can help the protein do its particular job, for instance grabbing a hydrophobic substrate in order to modify it chemically.
| Primary Structure | Secondary Structure |
|
|
| Tertiary Structure | Quaternary Structure |
|

|
| Figure 9: The various protein structures. | |
Depending on their R chains, amino acids also can vary in terms of their acidity and alkalinity. When the environment is neutral (pH 7), the amino acids aspartate and glutamate act as acids, whereas arginine and lysine act as bases, and this too has major implications for a protein’s properties.
Higher orders of protein structure
Except for very short chains (so short that they are usually not even called proteins), polypeptides bend and twist into complex shapes almost as soon as they are built, leading to secondary and tertiary protein structure. Secondary structure refers to any of a handful of regular shapes or patterns that form as a direct result of the primary structure, largely through a force called hydrogen bonding.
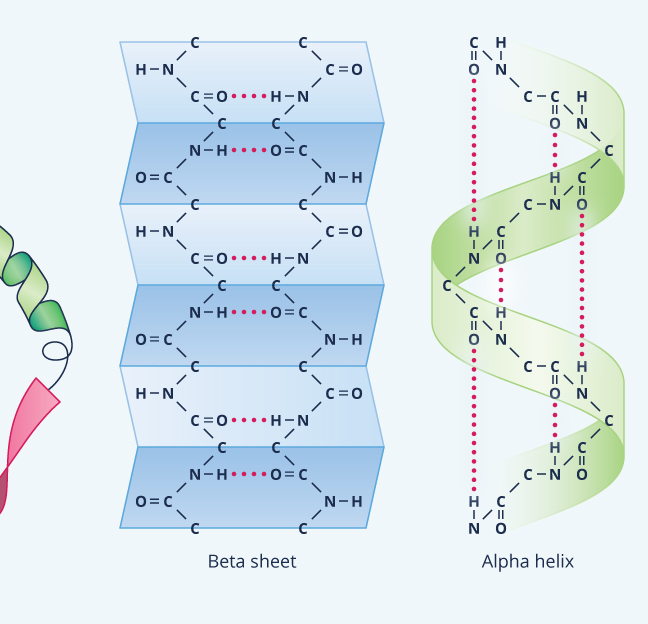
The most common secondary structure is an alpha helix (Figure 9). Think of it as kind of spiraling staircase. Each turn of the spiral consisting of 3.6 amino acids; in other words, four amino acids comprise more than one turn. Typically an alpha helix contains about 10 amino acids and thus three turns, but they also can be shorter or longer than this. As for their function, an alpha helix can provide shape as well as springy flexibility to the next level of protein structure, the tertiary structure. Consequently, they’re present in many different proteins, even small ones like insulin.
Another common secondary structure is called a beta-sheet, which forms when hydrogen bonds pull various non-adjacent segments, or “beta strands,” of polypeptide chain close together so that the primary structure folds back on itself multiple times (see Figure 9). The result is a ribbon-shaped area, which, like a helix, tends to stiffen and strengthen the protein.
Big proteins typically contain both alpha helices and beta-sheets. The small protein insulin helps to regulate the movement of glucose from the blood into cells by controlling the activity of another protein, the enzyme hexokinase. Unlike insulin, however, hexokinase is huge. Built of more than 900 amino acids, hexokinase has a good mix of both alpha-helices and beta-sheets. Hemoglobin, on the other hand, is almost completely alpha-helical, and antibodies consist almost completely of beta-sheets.
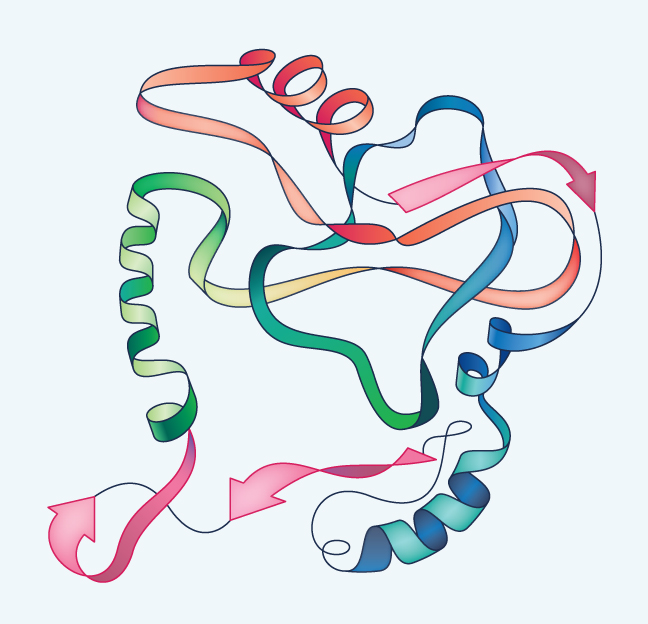
The presence of alpha-helices and beta-sheets, plus interactions between various amino acids not adjacent to one another in the chain, causes the protein to fold and twist still more, but in unique and irregular ways. This is the tertiary structure and it is stabilized not only by the alpha-helices and beta sheets within it, but often also by disulfide (S-S) bridges between cysteines. In explaining the structure of insulin, Sanger found one such S-S bridge contributing to the tertiary structure by connecting two cysteines that are both in the A chain but are not next to one another in the primary amino acid sequence. He also found two other S-S bridges connecting the A chain with the B chain.
Over the years, researchers found that large proteins typically contain many disulfide bridges. Lysozyme, for instance, an enzyme that immune cells use to destroy bacteria, has four disulfide bridges and antibodies have a different amount, depending on the antibody subtype. During the early 1970s, Argentinian researcher César Milstein helped to determine that disulfide bridges in antibodies are arranged in a particular pattern, a pattern that enables each antibody to take on the unique antibody shape (Figure 10). (See our profile César Milstein: Hybridoma Cells to Create Monoclonal Antibodies for more information on Milstein's research.) Disulfide bridges, however are not universal. Hemoglobin and a related protein called myoglobin, for instance, are famous for having no S-S bonds at all.
Figure 10: Schematic diagram of an antibody and antigens.
The final level of structure is quaternary structure (see Figure 9), which exists when two or more polypeptide chains come together. An example is hemoglobin, which consists of four chains. In addition to simply making the molecule big, the four chains of hemoglobin actually influence one another, causing an effect that helps the molecule to grab onto oxygen when blood circulates through the lungs and then give up the oxygen to tissues deep in the body where it is needed.
Not all proteins have a quaternary structure since many proteins consist of just one chain. Although Sanger found that insulin consisted of two chains, those two chains and the disulfide bonds connecting them are actually part of the tertiary structure, not quaternary. The reason is that insulin is made from a larger protein precursor called proinsulin in which chains A and B are connected by a third sequence, chain C. Rather than being made from separate chains, proinsulin is synthesized in cells as just one chain. The chain then bends on itself and the three disulfide bonds help in that process, but then the C chain is snipped out (Figure 11).
Figure 11: Structure of proinsulin showing C-peptide and the A and B chains of insulin.
image ©ZapyonComprehension Checkpoint
Protein structures in medical research
As in Frederick Sanger’s era, research on protein structure today has major implications in clinical medicine. Regarding secondary structure, for instance, researchers are developing an ability to detect and control certain diseases in their early stages. While beta-sheets are normal in many proteins, in some cases they are a sign of disease. A notable example is a substance called beta-amyloid, which is made when an otherwise normal protein in body cells develops beta-sheets that it’s not supposed to have. In Alzheimer disease, beta-amyloid accumulates within brain cells, leading to dementia and physical decline. It’s controversial, but scientists also suspect that beta-amyloid also accumulates in an aging brain in the absence of Alzheimer disease.
Recently, scientists at the University of Washington learned to synthesize an alternative secondary structure within proteins, called alpha-sheet. It is similar to the more common beta-sheet, except that it is flipped geometrically, kind of like a mirror image. Essentially the opposite of a beta-sheet, alpha-sheets can act as detectors for beta-sheets, similar to how a right hand can be used to "detect" the presence of a left hand in the dark. Not only do the researchers expect that proteins synthesized to contain alpha-sheets can be used for early detection of amyloid diseases, but the alpha-sheets also could be used for treatment in the form of drugs consisting of proteins with alpha sheets. Once in contact with the pathological beta-amyloid, such alpha-sheet drugs should disrupt the hydrogen bonding of the abnormal beta-sheets, thereby causing the beta-amyloid to revert back into a normal protein. That could be extremely helpful for individuals plagued with degenerative conditions such as Alzheimer disease, and possibly it may also open a new age of intervention against a more mild, but nevertheless damaging, process that traditionally has been dismissed inevitable for those reaching old age.